Evo 2 Is the Largest Open-Source Biology AI Ever Built
Arc Institute and NVIDIA release Evo 2, a 40B-parameter open-source AI trained on 9.3 trillion nucleotides from every domain of life, with full weights, code, and training data.
Arc Institute and NVIDIA published the Evo 2 paper in Nature today, and they dropped everything with it: full model weights, training code, inference code, and the 8.8 trillion-token pretraining dataset. Evo 2 is trained on 9.3 trillion nucleotides from over 128,000 whole genomes spanning every domain of life - bacteria, archaea, and eukaryotes including humans. The flagship is 40 billion parameters with a 1 million nucleotide context window.
It isn't the biggest model by parameter count. But it's the largest open-source AI ever released specifically for genomics, and one of the most fully open scientific foundation model releases in any domain.
Key Specs
| Spec | Value |
|---|---|
| Flagship size | 40B parameters |
| Also available | 20B, 7B, 1B |
| Context window | 1 million nucleotides |
| Training data | 9.3T nucleotides from 128K+ genomes |
| Architecture | StripedHyena 2 |
| Training hardware | 2,000+ NVIDIA H100 GPUs on AWS |
| License | Open source (weights + code + data) |
| Access | HuggingFace, NVIDIA BioNeMo, NIM |
The Model Family
Four Sizes for Different Hardware Realities
Evo 2 ships in four variants. The 40B flagship needs multiple NVIDIA Hopper GPUs with FP8 support via Transformer Engine. The 7B runs in bfloat16 on a single GPU with a standard pip install. The 1B base model targets researchers with minimal compute, at a 8K context. The 20B sits in between, released in February 2026 ahead of today's Nature paper.
That hardware split matters. The 7B is immediately accessible to any lab with a decent workstation. The 40B is for production-scale variant analysis at institutions with real GPU budgets.
StripedHyena 2 - Not a Transformer
The architecture is called StripedHyena 2, a hybrid Mamba-style state space model. Greg Brockman, OpenAI's co-founder and president, spent part of a recent sabbatical contributing to the design. The core property it buys: near-linear memory scaling with sequence length. Standard transformer attention scales quadratically with context - you can't run it at 1 million tokens without enormous hardware overhead. StripedHyena 2 processes sequences at single-nucleotide resolution and handles 8 times as many nucleotides per inference as Evo 1.
The pretraining dataset, OpenGenome2, contains 8.8 trillion tokens from all three domains of life - 30 times more data than Evo 1. Training ran for several months on 2,000+ NVIDIA H100 GPUs via NVIDIA DGX Cloud on AWS.
 Arc Institute's visualization of Evo 2's training coverage: the tree of life encoded as nucleotide sequences, from single-celled microbes to humans.
Arc Institute's visualization of Evo 2's training coverage: the tree of life encoded as nucleotide sequences, from single-celled microbes to humans.
What It Actually Does
Predicting Mutations Before They Kill
The clearest benchmark in the paper is BRCA1 zero-shot variant effect prediction. Evo 2 classifies over 90% of BRCA1 mutations correctly as benign or pathogenic, without task-specific fine-tuning or labeled training examples. BRCA1 has thousands of known variants with different clinical outcomes. Getting most of them right from sequence alone - no wet lab, no labels - is the kind of result that makes clinical researchers pay attention.
The model extends this to other genes and to noncoding regions, where most variant effect prediction tools historically fail. Noncoding mutations cause disease; they just lack the clear protein-change signal that makes coding mutations interpretable.
Designing Whole Genomes from Scratch
Beyond reading sequences, Evo 2 can write them. Arc demonstrated the model designing novel genomes comparable in length to simple bacterial genomes. Not editing existing sequences or making point mutations - creating plausible biological code from scratch. For synthetic biology, that's a meaningfully different capability than anything in prior open tooling.
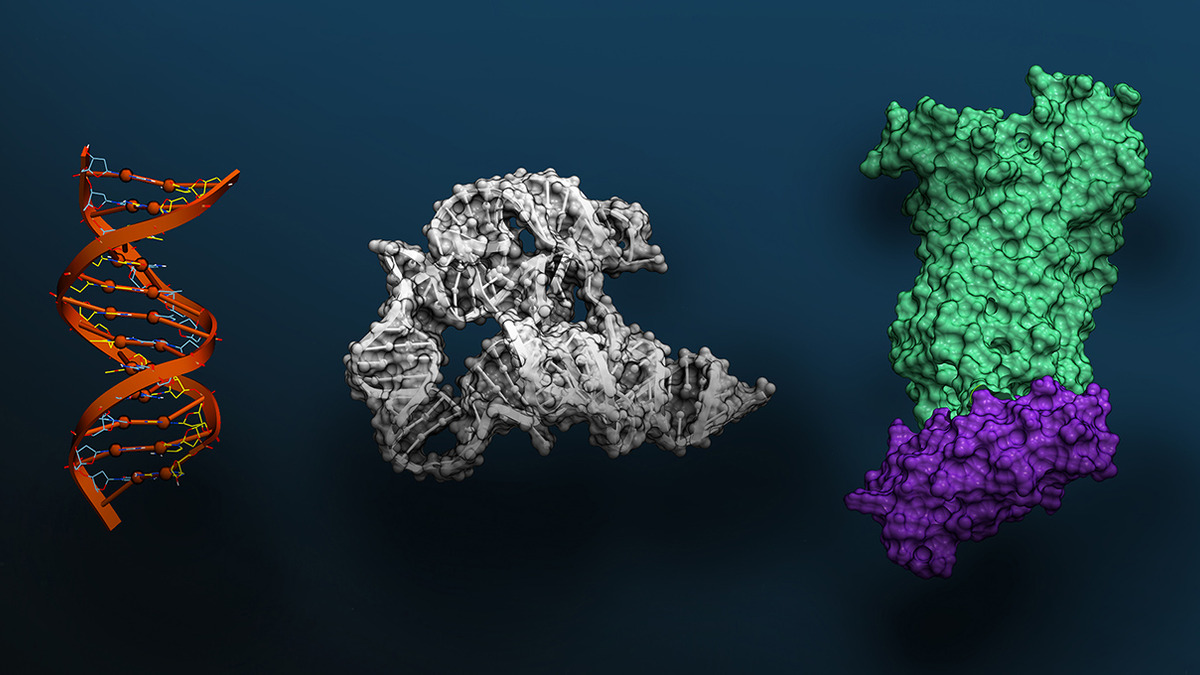 NVIDIA visualization of the three molecular scales Evo 2 models: DNA double helix, RNA tertiary structure, and protein surface topology.
NVIDIA visualization of the three molecular scales Evo 2 models: DNA double helix, RNA tertiary structure, and protein surface topology.
Benchmarks vs. Evo 1
| Capability | Evo 2 | Evo 1 |
|---|---|---|
| BRCA1 mutation classification | >90% accuracy | Lower baseline |
| Nucleotides per inference | 8x more | - |
| Training data scale | 9.3T tokens | ~300B tokens |
| Genome design | Full bacterial-scale | Fragment-scale only |
| Context window | 1M nucleotides | 8K nucleotides |
| Domains covered | Bacteria, archaea, eukaryotes | Primarily bacteria |
For context in the bioAI space: Isomorphic Labs' IsoDDE platform doubles AlphaFold 3's accuracy in protein structure prediction but keeps its weights completely closed. Evo 2 is explicitly positioned as the open alternative - it trades some peak benchmark performance for full reproducibility and community access.
Open Stack, Not Just Open Weights
Most model releases called "open" give you weights and nothing else. Arc is releasing:
- Model weights (all four sizes) on HuggingFace under the
arcinstituteorg - Training code via the GitHub repository
- Inference code with light and full install paths
- The full OpenGenome2 pretraining dataset
NVIDIA BioNeMo hosts the model for cloud inference, and NVIDIA NIM microservices handle containerized self-deployment. Arc also shipped two companion tools: Evo Designer, a web interface for direct interaction, and a mechanistic interpretability visualizer built with Goodfire that shows which genomic features the model attends to.
"Evo 2 represents a major milestone for generative genomics. By advancing our understanding of these fundamental building blocks of life, we can pursue solutions in healthcare and environmental science that are unimaginable today." - Patrick Hsu, Co-Founder, Arc Institute
This full-stack release puts Evo 2 in different territory than most academic model drops. It is closer to what NVIDIA did with Alpamayo, its open robotics model that shipped with full deployment infrastructure - than to the research-paper-with-partial-weights pattern common in bioAI.
Safety Guardrails
One design decision worth flagging: the training data explicitly excludes human-infecting pathogens. Arc's stated reason is biosecurity - preventing the model from being used to engineer dangerous viruses. That exclusion is a real limitation for virologists studying pathogen genomes. Whether the exclusion is solid enough in practice against adversarial inputs is a question the paper doesn't fully address.
What To Watch
The Eukaryote Gap
Most of the training data is microbial. Bacteria and archaea have compact, well-annotated genomes that are easier to learn from. Human and plant genomes are messier - large noncoding regions, alternative splicing, complex regulatory architecture. Evo 2's benchmarks lean bacterial. Performance on complex human gene regulation in novel contexts still needs independent validation.
Compute Access at Scale
The 7B model is truly accessible. The 40B isn't - multiple H100 GPUs means a hardware cost that most academic labs can't absorb. NVIDIA BioNeMo provides a cloud path, but that reintroduces cost and dependency. The open weights are real, but operational access at scale still requires Nvidia-class infrastructure.
Independent Validation
Evo 2 published today. The first wave of tests from labs outside Arc's collaboration - Stanford, Berkeley, UCSF - will show whether the benchmark numbers hold on novel datasets, whether designed genomes are biologically viable in lab conditions, and whether the tool integrates cleanly into standard bioinformatics pipelines.
"Evolution has left its imprint on biological sequences, with patterns refined over millions of years containing signals about molecular function," said Brian Hie, a Stanford professor involved in the research. Evo 2 is the most serious attempt yet to read those patterns at scale, in the open, with the full stack available for anyone to run.
Sources:
